Chen's Reaserch Lab
Open Source
iShapEditing: Intelligent Shape Editing with Diffusion Models[Project Code]We introduce iShapEditing, a novel framework for 3D shape editing which is applicable to both generated and real shapes. Users manipulate shapes by dragging handle points to corresponding targets, offering an intuitive and intelligent editing interface. Leveraging the Triplane Diffusion model and robust intermediate feature correspondence, our framework utilizes classifier guidance to adjust noise representations during sampling process, ensuring alignment with user expectations while preserving plausibility. For real shapes, we employ shape predictions at each time step alongside a DDPM-based inversion algorithm to derive their latent codes, facilitating seamless editing. iShapEditing provides effective and intelligent control over shapes without the need for additional model training or fine-tuning. Experimental examples demonstrate the effectiveness and superiority of our method in terms of editing accuracy and plausibility. 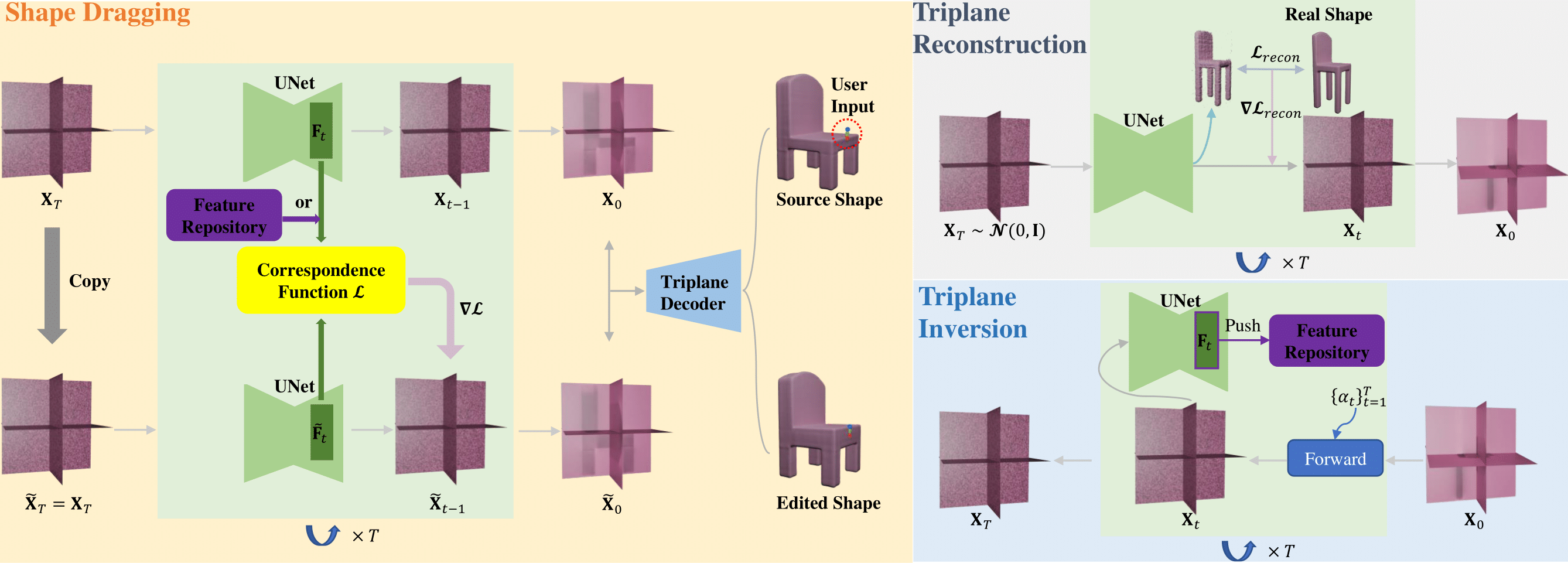
|
DTGBrepGen: A Novel B-rep Generative Model through Decoupling Topology and Geometry[Project Code]Boundary representation (B-rep) of geometric models is a fundamental format in Computer-Aided Design (CAD). However, automatically generating valid and high-quality B-rep models remains challenging due to the complex interdependence between the topology and geometry of the models. Existing methods tend to prioritize geometric representation while giving insufficient attention to topological constraints, making it difficult to maintain structural validity and geometric accuracy. In this paper, we propose DTGBrepGen, a novel topology-geometry decoupled framework for B-rep generation that explicitly addresses both aspects. Our approach first generates valid topological structures through a two-stage process that independently models edge-face and edge-vertex adjacency relationships. Subsequently, we employ Transformer-based diffusion models for sequential geometry generation, progressively generating vertex coordinates, followed by edge geometries and face geometries which are represented as B-splines. Extensive experiments on diverse CAD datasets show that DTGBrepGen significantly outperforms existing methods in both topological validity and geometric accuracy, achieving higher validity rates and producing more diverse and realistic B-reps. 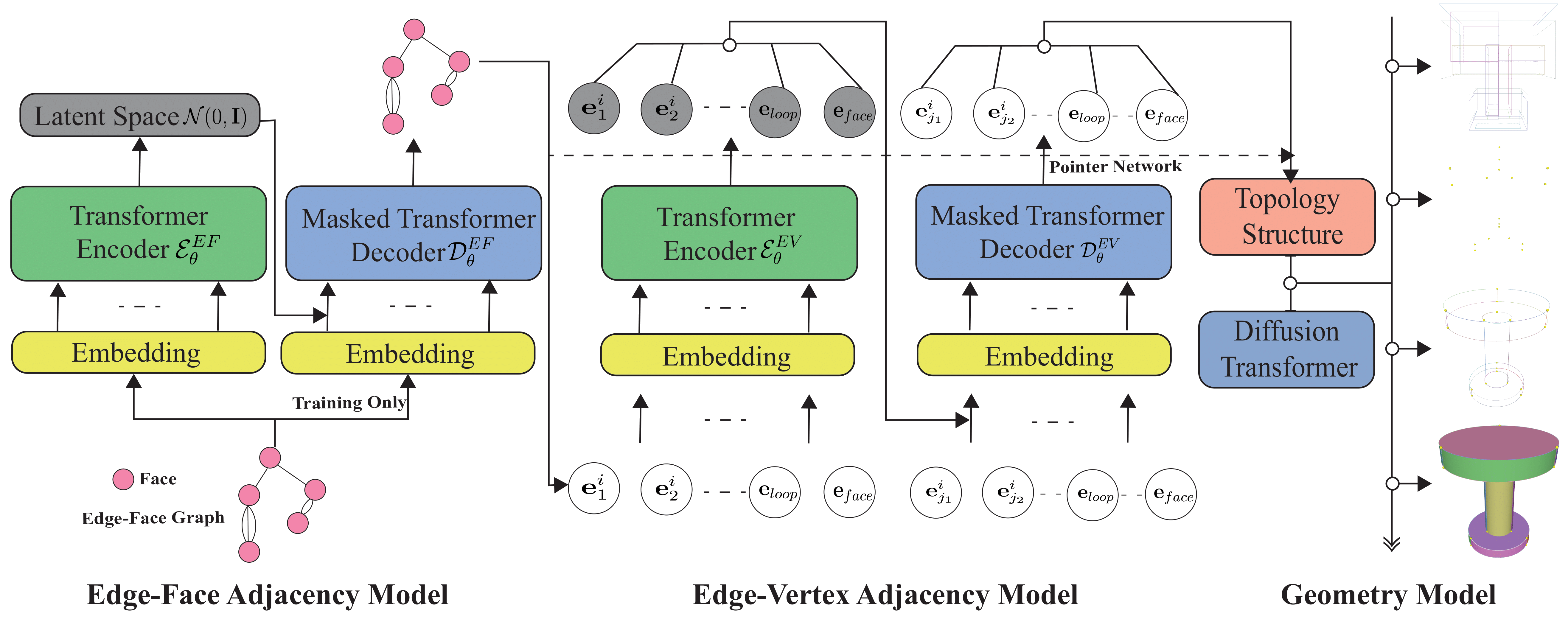
|
WEDGE: imputation of gene expression values from single-cell RNA-seq datasets using biased matrix decomposition[Project Code]The low capture rate of expressed RNAs from single-cell sequencing technology is one of the major obstacles to downstream functional genomics analyses. Recently, a number of imputation methods have emerged for single-cell transcriptome data, however, recovering missing values in very sparse expression matrices remains a substantial challenge. Here, we propose a new algorithm, WEDGE (WEighted Decomposition of Gene Expression), to impute gene expression matrices by using a biased low-rank matrix decomposition method. WEDGE successfully recovered expression matrices, reproduced the cell-wise and gene-wise correlations and improved the clustering of cells, performing impressively for applications with sparse datasets. Overall, this study shows a potent approach for imputing sparse expression matrix data, and our WEDGE algorithm should help many researchers to more profitably explore the biological meanings embedded in their single-cell RNA sequencing datasets. 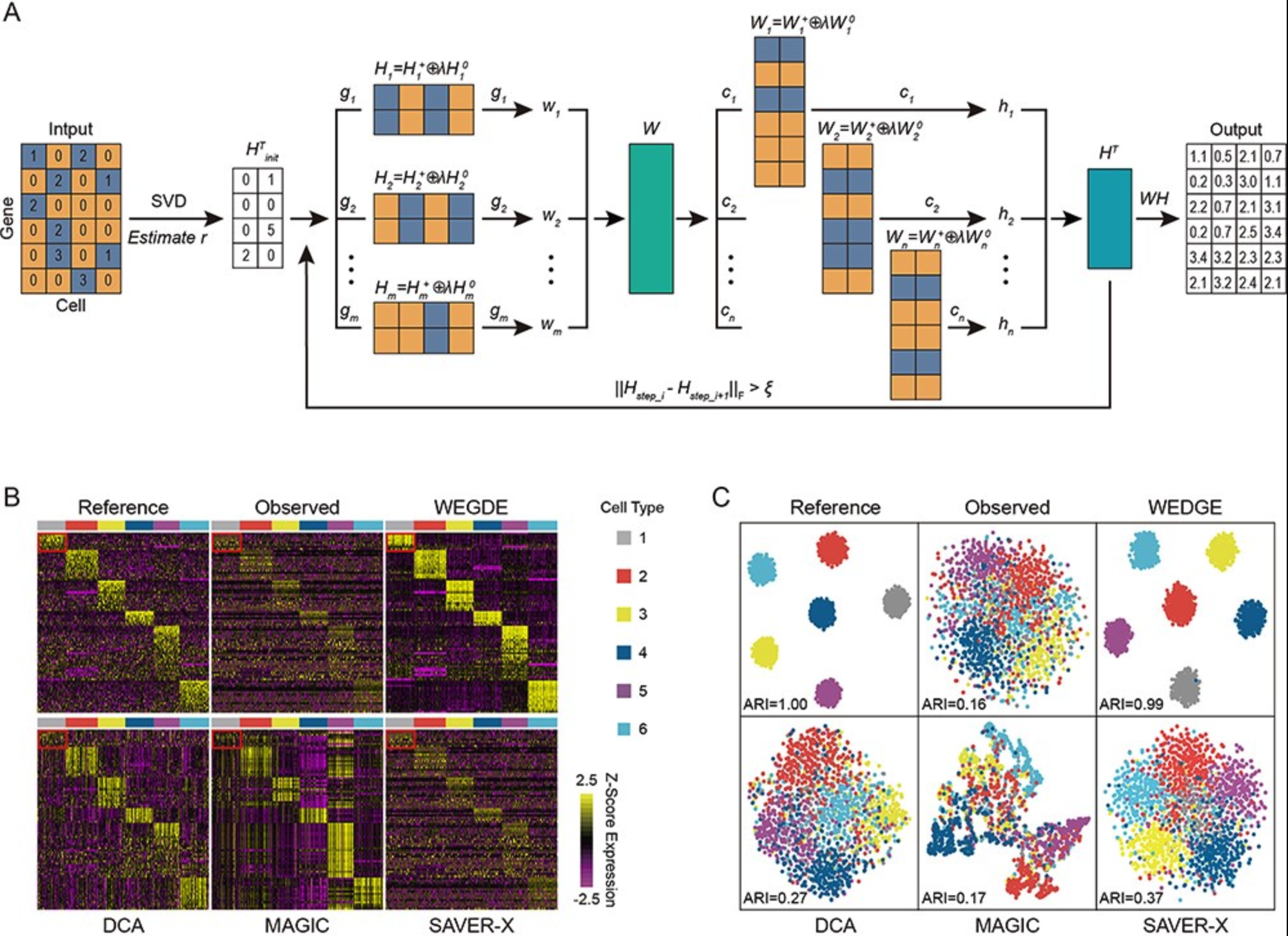
|
Benchmarking algorithms for single-cell multi-omics prediction and integration[Project Code]The development of single-cell multi-omics technology has greatly enhanced our understanding of biology, and in parallel, numerous algorithms have been proposed to predict the protein abundance and/or chromatin accessibility of cells from single-cell transcriptomic information and to integrate various types of single-cell multi-omics data. However, few studies have systematically compared and evaluated the performance of these algorithms. Here, we present a benchmark study of 14 protein abundance/chromatin accessibility prediction algorithms and 18 single-cell multi-omics integration algorithms using 47 single-cell multi-omics datasets. Our benchmark study showed overall totalVI and scArches outperformed the other algorithms for predicting protein abundance, and LS_Lab was the top-performing algorithm for the prediction of chromatin accessibility in most cases. Seurat, MOJITOO and scAI emerge as leading algorithms for vertical integration, whereas totalVI and UINMF excel beyond their counterparts in both horizontal and mosaic integration scenarios. Additionally, we provide a pipeline to assist researchers in selecting the optimal multi-omics prediction and integration algorithm. 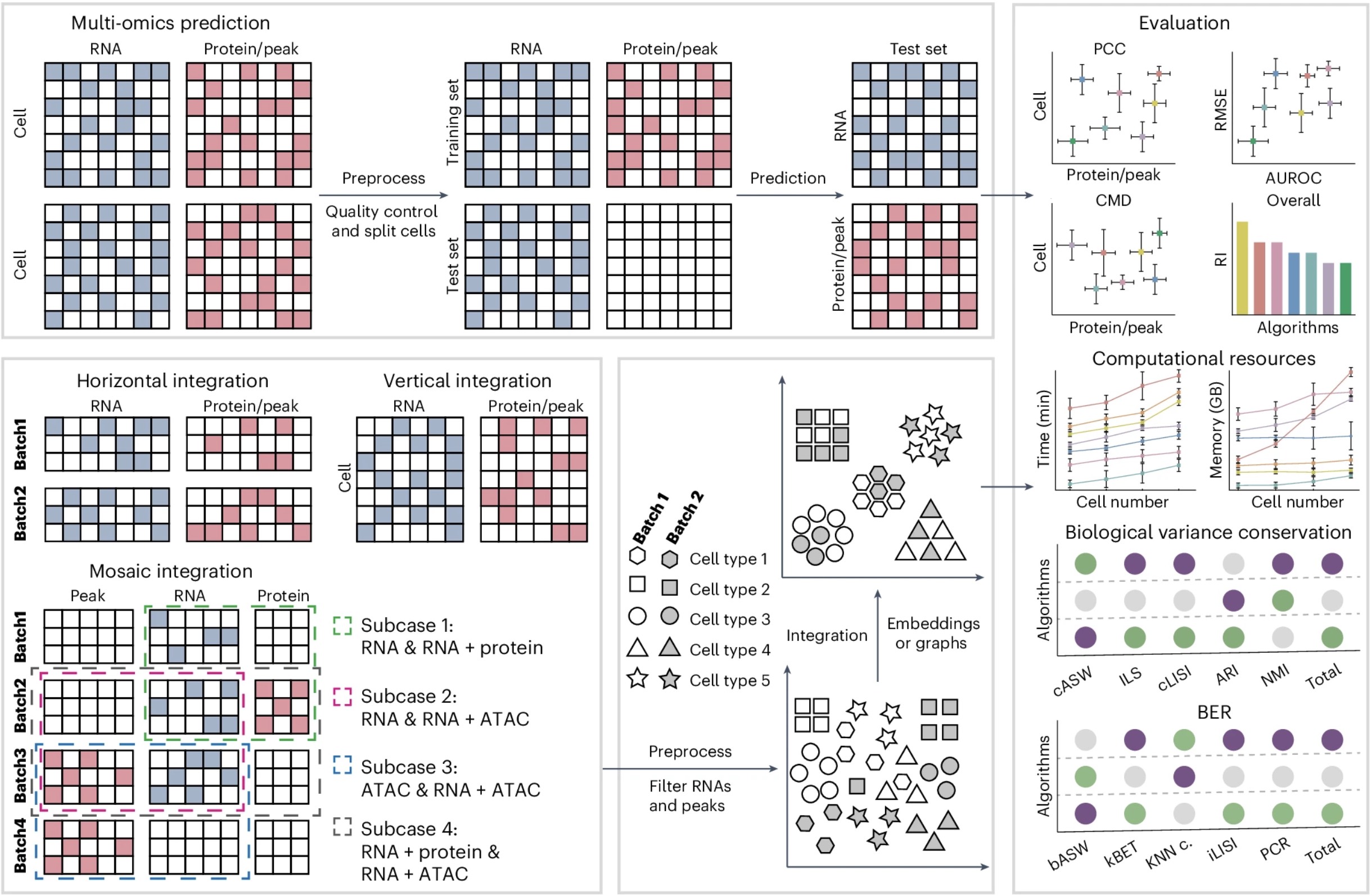
|